��Ë@��̖��F�}�j���A�� �@�QMS�EGVP�����̍s�����m�ɂ���Ë@�틖�F���E���k�T�C�g

|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
�@ �@
�@
�@ |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
ver2.0�i���i��Ë@�퓙�@�Ή��j �@
�@ 2014�N11��25���Ɉ��i��Ë@�퓙�@�i�u���i�A��Ë@�퓙�̕i���A�L�����y�ш��S���̊m�ۓ��Ɋւ���@���v�j���{�s����܂����B ���̖@���͏]���́u�@�v�����e�Ɩ��̂�ύX���ꂽ���̂ł��B����ɂ��A��Ë@��Ɋւ��鋖�F���傫���ς��܂����B
|

|
��Ë@��Ƃ́A���̂悤�Ȃ��̂������܂��B ���i��Ë@�퓙�@�i���̖@�j��2���4�� ��Ë@�����̂��̂⎡�ÖړI�̂��̂Ɍ���Ȃ����Ƃɒ��ӂ��ĉ������B
�@ |
 ���X�A���ˊ�A��×p�K�[�[�Ȃǂ���A��pX���f�f���u�Ȃǂ̈�p�d�C�@��A���͋@��܂ŁA��Ë@��ɂ́A���܂��܂Ȍ`��A�p�r�̂��̂�����܂��B
���X�A���ˊ�A��×p�K�[�[�Ȃǂ���A��pX���f�f���u�Ȃǂ̈�p�d�C�@��A���͋@��܂ŁA��Ë@��ɂ́A���܂��܂Ȍ`��A�p�r�̂��̂�����܂��B
 ��Ë@��̐����̔��ɂ́A�����Ǘ��E�i���Ǘ��E���S�Ǘ��̐ӔC�����߂���
��Ë@��̐����̔��ɂ́A�����Ǘ��E�i���Ǘ��E���S�Ǘ��̐ӔC�����߂���
|
��Ë@������А��i�Ƃ��Ďs��ɗ��ʂ����邱�Ƃ��A��s�Ƃ�������A�����̔��Ƃ������肵�܂��B �����̔����闧��̉�Ђ́u��Ë@�퐻���̔��Ǝҁv�Ƃ����܂��B�����̔����A�����Ƃ����邱�Ƃ�����܂��B ���ҁA�g�p�҂̈��S�����A�܂��A��Ï�̗L������S�ۂ��邽�߁A��@�@�i���i��Ë@�퓙�@�j�́A��Ë@����̔������Ђɑ��A��Ë@����i����ۏ����A���S���Ǘ�����ӔC�����߂Ă��܂��B ��Ë@��̐����̔��̂��߂ɂ́A���Ǝ҂��u��Ë@�퐻���̔��Ƌ����v�̎擾���K�{�ł��B QMS�ȗ��Ƃ�����ɏ]���Đ����Ǘ��E�i���Ǘ����s���AGVP�Ƃ�����Ɋ�Â����S�Ǘ����ł���̐��������Ă��邱�Ƃ��A���擾�̗v���ɂȂ��Ă��܂��B �����̔��Ǝ҂́A���琻�����������Ă��邱�Ƃ�����܂����A�K���������А����ł���K�v�͂���܂���B �������E�H�ꂪ�q��Ёi�ʖ@�l�j�ł����Ă��悢�ł����A���̋Z�p���������Ǝ҂ɐ����ϑ����邱�Ƃ��\�ł��B
|
��Ë@��̐����s�ׂɂ͐����Ɠo�^���K�v
|
��Ë@������鎖�Ǝ҂̂����A�u�v�v(*)�A�u�傽��g���v�u�ŋہv�u�ŏI���i�̕ۊǁv���s�����Ǝ҂́A�u��Ë@�퐻���Ɠo�^�v���K�v�ł��B ���Ə��������ł���ꍇ�͓��R�ł����A�O������̗A����A�O���Őv���Ă���悤�ȏꍇ�ɂ́A���Y�O���̎��Ə��ɂ��āA�u�O�������Ǝғo�^�v���K�v�ł��B �o�^�̎擾�́A�v�������ӔC�ҁu�ӔC�Z�p���v�����n�Ǘ��ł���悤�z�u���邱�Ɠ����v���ƂȂ��Ă��܂��B �\���ݔ��̓K���́A�o�^�v���ɂ͂Ȃ��Ă��܂���B(**) ���������āA���@���ォ�猩��A��Ë@�퐻���Ɠo�^�͎擾���₷���Ȃ����ƌ�����ł��傤�B �������A��Ë@��̐����ɂ������āA���̈�Ë@��̐����Ǘ��̂��߂ɕK�v�Ȑݔ��A���A��Ɗ��������߂��邱�Ƃɕω��͂���܂���B�ݔ����A��Ɗ����ɂ��ẮAQMS�ȗ߂Ƃ��������Ǘ��E�i���Ǘ��̊�̒��ɋK�肪����A�����Ƃ��Ă���ɏ]���ĊǗ�����邱�ƂɂȂ�܂��B �i�ڍׂ�6�����������
>>�j
�@ (*)�N���X�P��Ë@��̐v�݂̂��s���̂ł���A�����Ɠo�^�͕s�v�ł��B�܂��A�����̔��Ƌ��̎��Ə��Őv���s���ꍇ���A�o�^�s�v�ł��B (**) ���@�ł́A��Ë@�퐻���Ƌ��i�O����Ë@�퐻���ƎҔF��j�Ƃ������x�ł������A�\���ݔ��̗v�����ɘa����܂����B �@ �@ |
��Ë@���̔��A���n�A�ƂƂ��đݗ^����ꍇ�ɂ������K�v�ł�
|
��Ë@�퐻���̔��Ƌ��́A�����̔��Ǝ҂Ƃ��Ĉ�Ë@����s��ɗ��ʂ����邽�߂ɕK�v�ȋ��ł����A ��Ë@��̐����̔������ǂ��ł��邩�ɂ�����炸�A���X�N�̍����u���x�Ǘ���Ë@��v�Ɏw�肳��Ă����Ë@���A�ێ�_���ɐ��I�Ȓm����Z�\��K�v�Ƃ���u����ێ�Ǘ���Ë@��v���A��ʃ��[�U�[�ɔ̔���������n�A���݂����肷��ꍇ�ɂ́A�u���x�Ǘ���Ë@�퓙�̔��Ɓi�ݗ^�Ɓj�����v���K�v�ł��B �܂��A���X�N����r�I�Ⴂ�u�Ǘ���Ë@��v����ʃ��[�U�[�ɔ̔�����ɂ́A�u�Ǘ���Ë@��̔��Ɓi�ݗ^�Ɓj�́v���K�v�ł��B ��Ë@������Ж��`�Ő����̔����邽�߂ɂ́A�����̋Ƌ��E�F�肪�K�v�Ȃق��A ��Ë@�펩�̂��A��Ë@��ɗv������Ă�����{�v���A�i�ڌŗL�̗v������(JIS��IEC�Ȃǂ����p����Ă���Ⴊ�����ł��j�ւ̓K�����K�v�ł��B �@
|
�@
�@
![]() �Q
��Ë@���"�N���X"���m�F���܂��傤
�Q
��Ë@���"�N���X"���m�F���܂��傤
��Ë@��̃��X�N�ɉ������u�N���X���ށv�Ƃ�
|
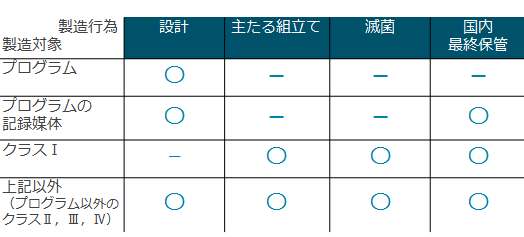
��Ë@��i��×p��j�ɂ́A�l�́i�������܂ށj�ɒ��ڐG�����́A�̓��ɗ��u������́A�����E���͗p�̂��̂Ȃǂ��܂��܂ȗp�r�̂��̂�����A�l�̓��ɑ���댯�x���قȂ�܂��B ��Ë@��́A���i�̃��X�N�ɂ��A�N���X�P�i��j�`�N���X�S�i���j�܂ł̂S��ނɕ��ނ���Ă��܂��B
|
![]() �N���X���ޕ\�Ŋm�F
�N���X���ޕ\�Ŋm�F
|
��Ë@����̔�������A�̔��E���ݓ��������肷��ɂ������ẮA�N���X�ɂ���āA�K�v�ƂȂ鋖��葱�����قȂ�܂��B ���������āA�܂��A�戵�\��̈�Ë@��̃N���X���ނ��m�F����K�v������܂��B �܂��A�댯�x�͒Ⴍ�Ă��ێ�Ǘ��ɐ��m�����K�v�Ȉ�Ë@��́A�u����ێ�Ǘ���Ë@��v�Ɏw�肳��Ă��܂��B�����̈�Ë@��i��×p��j�́A�����������u�N���X���ޕ\�v�Ɍf�ڂ���Ă��܂��B �������Ƃ����Ë@��̍\���A�g�p�ړI�A���\���ʁA�ȂǂƁA�N���X���ޕ\�̗ޕʁA��`�Ƃ��ƍ����ĉ������B ��ʓI���̂��ƂɁA�F�؊�E���F��E���F�R���K�C�h���C������߂��Ă��邱�Ƃ�����܂��̂ŁA �@ �@�@ �N���X���ޕ\�ɏo�Ă�����̂�������Ë@�킾�Ƃ������Ƃł͂���܂���B �������Ƃ����Ë@�킪�N���X���ޕ\�̈�ʓI���̂̒�`�ɊY�����Ȃ��ꍇ�́A������u�V��Ë@��v�i�V�K���̂����Ë@��j�ɊY������ꍇ������܂��B ��Ë@��ɊY�����邩�ǂ����́A�@��2���4���ɂ��ƂÂ��A�\���A�@�\�A�g�p�ړI�Ȃǂ��瑍���I�ɔ��f���܂��B �@
�@ |
![]() �R
��@�@���F�̎d�g�݂̐��x�E���K���𗝉����܂��傤
�R
��@�@���F�̎d�g�݂̐��x�E���K���𗝉����܂��傤
���Ђň�Ë@����i�����ϑ�����ꍇ���܂ށj������A�A�������肵�āA���Ђ̖��O�Ŏs��ɗ��ʂ�����i�����̔��j�ɂ́A���i��Ë@�퓙�@�Ɋ�Â��u��Ë@�퐻���̔��Ƌ����v���K�v�ł��B
��Ë@�퐻���̔��Ƌ��́A�戵��Ë@��̃N���X�ɉ����đ�P�`�R��ɕ�����Ă��܂��B
�܂��A�N���X�R�A�S�ɊY�������Ë@���A����ێ�Ǘ���Ë@����A��Ƃ�[�U���ɔ̔��E���n�E���݂���ɂ́A��Ë@�퐻���̔��Ƌ��Ƃ͕ʂɁA�u���x�Ǘ���Ë@�퓙�̔��ƁE���Ƌ����v���K�v�ł��B
�i���x�Ǘ���Ë@��̔��ƁE���Ƌ��������Ă���̔��ƎҁA�����̔��ƎҁA�����Ǝ҂����ɏ��n����ꍇ�͕s�v�j
�N���X�Q�̈�Ë@��̔̔��i����ێ�Ǘ���Ë@��ȊO�j�̔̔��ɂ́A�u�Ǘ���Ë@��̔��Ɠ́v���K�v�ł��B
�܂��A��Ë@��̐����H���̂������̍H����S�����鐻�����́A��Ë@�퐻���Ɠo�^���K�v�ł��B
�@
�@��Ë@����̔��i�A�����܂ށj����ꍇ�̋K��
��Ë@��̃N���X�ɉ����ĕK�v�ȋ����قȂ�܂��B
|
�K���Ώ� |
�����̔��Ǝ� |
��Ë@��i���i�j |
�̔��X�E�c�Ə� |
|
��Ë@�� |
|||
|
�N���X�V�A�W |
��P�� |
|
|
|
�N���X�U |
��Q�� |
���F |
�Ǘ���Ë@�� |
|
|
��Q�� |
�F�� |
�Ǘ���Ë@�� |
|
�N���X�T |
��R�� |
|
�| |
��P���Ë@�퐻���̔��Ƌ����擾����ƁA�N���X�R�A�S�����łȂ����ʂ̃N���X�i�P�C�Q�j�̈�Ë@��������̔��\�ł��B
��Q���Ë@�퐻���̔��Ƌ����擾����ƁA�N���X�P�̈�Ë@��������̔��\�ł��B
�i���j�F�؊�E�E�E�@�u�w��Ǘ���Ë@��v�́u�K�����F�؊�v�̂��ƁB
�@![]() �@�w��Ǘ���Ë@���i��錧���̃y�[�W�j
�@�w��Ǘ���Ë@���i��錧���̃y�[�W�j
�@![]() �@�F�؊�A�K�p�ƂȂ�JIS���̌����i���i��Ë@�푍���@�\�̃y�[�W�j
�@�F�؊�A�K�p�ƂȂ�JIS���̌����i���i��Ë@�푍���@�\�̃y�[�W�j
![]() �@
�@
�@
�@
�@��Ë@��̐������A�ۊǏꏊ���̋K��
��Ë@������鎖�Ə��́A�S������H���ɂ��A��Ë@�퐻���Ɠo�^�A�܂����O�������Ǝғo�^���K�v�ȏꍇ������܂��B
���@�ɔ�ׁA��������o�^���ɂȂ�A�܂��\���ݔ��v�����ɘa����铙�A�V�K�Q���͂��₷���Ȃ�܂����B
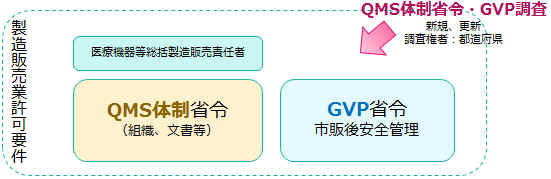
�����A�����Ǝ҂́AQMS�ȗ߂Ɋ�Â��������̕i���}�l�W�����g�V�X�e���iQMS�j���\�z����K�v������AQMS�̑̐��\�z�͐����̔��Ǝ҂ɂ����Ҋč���R���@�ւɂ��R���̑ΏۂƂȂ�܂��B
�\���ݔ��͓s���{���̐R�����鋖�v���ł͂Ȃ��Ȃ������̂́A��������QMS�ȗ߂̊֘A����K��Ɋ�Â��K�ɉ^�p�Ǘ�����Ȃ���Ȃ�܂���B
![]() �@��Ë@��̐����̔��ɂ��Ƃ߂��鐻���Ǘ��E�i���Ǘ��E���S�Ǘ��̐�
�@��Ë@��̐����̔��ɂ��Ƃ߂��鐻���Ǘ��E�i���Ǘ��E���S�Ǘ��̐�
��Ë@����s�ꗬ�ʂ����鎖�Ǝҁi�����̔��ƎҁB�A���Ǝ҂������Ɋ܂܂�܂��j�́A��Ë@��̐����̔��Ƌ����擾���Ȃ���Ȃ�܂���B
��Ë@�����{�����s��ɏ�s����ɂ������ẮA��Ë@��̕i����ۏ��A���[�U�[�⊳�ҁA��ÊW�ғ��̈��S���Ǘ�����ӔC���܂��B
���̂��߁A�@�ł́A�����̔��Ƌ��̗v���Ƃ��āA�����Ǘ��E�i���Ǘ������S�Ǘ��̑̐��𐮂��邱�Ƃ����߂Ă��܂��B
��Ë@�퐻���̔��Ƌ��͈�@�l�ɂЂƂ̋��ł��B
�i��1���Ë@�퐻���̔��Ƌ��Ƒ�3���Ë@�퐻���̔��Ƌ����Ɏ����Ƃ͂���܂���j
���������̔��ӔC�ҁi��q�j�̋Ζ����鎖�Ə��i�{�Г��j������s���{���m���ɁA����\�����܂��B
|
|
�@
![]() ��Ë@�퓙���������̔��ӔC���@[2012�N8��30���@�v������]
��Ë@�퓙���������̔��ӔC���@[2012�N8��30���@�v������]
��Ë@��̐����̔�������ӔC�҂ł��B
���L�̎��i�v�����������펞�z�u���Ă��������B
��Ŕz�u����B [���������̔��ӔC���̎��i�v���n
|
![]() �����i���Ɩ��^�c�ӔC��
�����i���Ɩ��^�c�ӔC��
�i���ۏؕ���̐ӔC�҂ł��B
|
[�����i���Ɩ��^�c�ӔC���̊�n |
�i�ӂɂ͈�Ë@��A���i���͍Đ���Ó����i���̋��Ǝ҂ɂ�����3�N�̕i���Ǘ����̎����o�������߂���_�ɗ��ӂ��Ă��������B�i�����3��ɂ�����o���͌����3��̐\�����ɂ̂ݔF�߂��܂��j
�m���i���X�N�����Ă��A�Ɩ��o�����\���L���铙�A�W�Ɩ����n�m�����҂ł���ׂ��n�Ƃ���Ă��܂��B
�@
�u���̑�����ɗނ���Ɩ��v�Ƃ́A
�@���������̔��ӔC��
�@�����Ǘ���
�@�A���Ǘ���
�@�i���Ǘ��ӔC��
�@�����Ǘ��ӔC��
���Ƃ���Ă��܂��B
��2��A��3��̏ꍇ�́A���̂悤�ȕ����܂܂�܂��B
�@ISO9001����13485�̔F���擾���Ă��鐻���Ǝҁi��Ë@��Ɍ���Ȃ��j�ɂ�����i���}�@�l�W�����g�V�X�e���̈ێ��Ǘ��̌o���A���͓����č��̌o���@3�N�ȏ�
![]() ���S�Ǘ��ӔC��
���S�Ǘ��ӔC��
���S�m�ۋƖ��̐ӔC�҂ł��B
|
[���S�Ǘ��ӔC���̊�n |
��1��̏ꍇ�́A���S�Ǘ����������u���A���̐ӔC�҂ł��邱�ƁA
�܂��A���S�m�ۋƖ���3�N�ȏ�]�������o�������߂��܂��B
��2��A��3��́A3�N�̌o���͋��߂��Ă��܂���B
�u���̑�����ɗނ���Ɩ��v�́A�s�̌㒲���ӔC�҂̋Ɩ�����������Ƃ���Ă��܂����A�����S���ҁA��Ë@����S���҂Ƃ��Ă̋Ɩ��݂̂ł͔F�߂��Ȃ��Ƃ���Ă��܂��B
![]() �����Ǘ��E�i���Ǘ��̐��A���S�Ǘ��̐�
�̍\�z�i�U�Q�Ɓj
�����Ǘ��E�i���Ǘ��̐��A���S�Ǘ��̐�
�̍\�z�i�U�Q�Ɓj
|
���́A�����Ǘ��E�i���Ǘ��̑̐��Ɋւ����iQMS�̐��ȗ��j�A��Ë@��̎s�̌���S�Ǘ��Ɋւ����iGVP�ȗ��j���A�ȗ߂Ƃ��Č��z���Ă��܂��B ��Ë@��̐����̔��Ƌ��Ǝ҂́A�����̏ȗ߂ɏ����āA�K�ɁA��Ë@��̐����Ǘ��E�i���Ǘ��̑̐������A�܂��AQMS�ȗ߁EGVP�ȗ߂Ɋ�Â��Ɩ����s��˂Ȃ�܂���B QMS�̐��EGVP�̐��������Ă��邱�Ƃ��A�u���̎擾�v�u���̈ێ��v�̗v���ł��B ��Ë@��̐����̔��ɂ����āA�ł���{�I�ȗp��ł��̂ŁA���Њo���Ă��������B �@ |
|
�����Ǘ��E�i���Ǘ��̊�iQMS�j�A���S�Ǘ��̊�iGVP�j�̉���y�[�W�ł� |
![]()
�@
�@�@
![]() �T
QMS�^GVP�̑̐��\�z�@�i�g�D�A�菇���A�L�^�ށj
�T
QMS�^GVP�̑̐��\�z�@�i�g�D�A�菇���A�L�^�ށj
![]() ��Ë@�퐻���̔��Ǝ҂́AQMS�AGVP�̑̐��\�z���K�{�v��
��Ë@�퐻���̔��Ǝ҂́AQMS�AGVP�̑̐��\�z���K�{�v��
|
��Ë@�퐻���̔����Ǝ҂́A���Ƃ̐��s�̂��߂ɁA�菇�������A�����E��E�o�ׁE��������������W�E�Ȃǂ̋L�^������ȂǁA�菇���ɂ��^�p�����邱�Ƃ��K�v�ł��B ��Ë@�퐻���̔����Ǝ҂́AQMS�̐��ȗ߁EQMS�ȗ��Ɋ�Â��A�u�i���Ǘ��ēV�X�e������v�≺�ʂ̊e�핶���A�L�^�ނ��쐬����`��������܂��B �܂��AGVP�ȗ��Ɋ�Â��A�s�̌���S�Ǘ��̐��𐮂��Ă����˂Ȃ�܂���B QMS�AGVP�̑̐��̐����͈�Ë@�퐻���̔��Ƃ́u���v���v�ɁAQMS�ւ̓K���͈�Ë@��̐����̔����F��F�̎擾�̗v���ɂȂ��Ă��܂�����A ��Ë@��̋Ƌ��\�����Ë@��̏��F�E�F�ؐ\���̂Ƃ��܂łɂ́A�菇����L�^�ށA�ӔC�҂�S���҂̌���A�u��̓I�ɍ�肱�菇���v�A�^�p�ł���̐��Â��肪�K�v�ł��B �@ |
|
�����Ǘ��E�i���Ǘ��̊�iQMS�j�A���S�Ǘ��̊�iGVP�j�̉���y�[�W�ł� |
![]()
�@�@�@
![]() QMS�EGVP�菇���́A���Ԃɍ��������̂𥥥
QMS�EGVP�菇���́A���Ԃɍ��������̂𥥥
�s���{���ł́A�R�����ɗp�ӂ���Ă���菇�����K�ł��邩�ǂ����A���̓��e�����Ǝ҂��������āA�K�ɉ^�p�ł�����ǂ����Ȃǂ��A���n�����Ŕ��f���܂��B �\���i�K�ł͂������܂����Ƃ̓X�^�[�g���Ă��܂��A��舵����Ë@��A��Ђ̑g�D�A�̘H���ɉ����A���擾��K�ɉ^�p�ł���悤�Ȏ菇���E�g�D�ɂȂ��Ă��邱�Ƃ��A�V�K�\�����̌��ł��B ���������̂����k�E���˗��Č��̒��ɂ́A����\�������ꂽ���Ǝ҂̂����ŁAQMS�EGVP�菇�����s�K���Ȃ��߂ɂȂ��Ȃ�������OK�����炦���A���擾���Ȃ��Ȃ��ł��Ȃ������������܂��B �Ȃ��Ȃ�OK���ł܂���ƁA�����\�肵�������Ɏ��Ƃ��X�^�[�g�ł��Ȃ��悤�Ȃ��Ƃɂ��Ȃ肩�˂܂���B �ł�����A�����i�K�ŁA����P����ʂ��āA��@�@���F��m��AQMS�EGVP�̗v���������悭�������āA �T�v���C���[�E�������Ƃ̊W�A������Ë@��̓����A�Г��g�D���܂��A���̂���菇�������邱�Ƃ�������K�{���Ƃ�����ł��傤�B ���������ł́A�V�K�\���⋖�X�V���ɍۂ��A����P���̈�Ƃ��āA����/���S���җl�ɖ@�EQMS�EGVP�������������������ƂɎ���u��������P���i�Г�����j�̋@���݂��Ă���܂��B
|
|
����������QMS�EGVP�菇���E�L�^�l�������̎x�����s���Ă���܂��B |
![]()
�@
�@
![]() �����X�V
�����X�V
|
��Ë@��̏��F�E�F���ێ����A�܂���Ë@�퐻���̔��Ƌ�����Ɠo�^���X�V���邽�߂ɂ́AQMS�AGVP�̐��̐�����QMS�ȗ߂ւ̓K���A�i�ڂ��Ƃ̋L�ڐ����Ȃǂ̏��葱�����K�v�ł��B ���߂Ă̋��擾���ƈقȂ�A�O��̍X�V�ȍ~��QMS�EGVP���K�ɉ^�p����Ă������ǂ���������܂��B �R���i�K��QMS�EGVP�̉^�p���\���łȂ��A�Ƃ����ɂȂ�܂��ƁA�ꍇ�ɂ���Ă͋��������߂��Ă��X�V�ł��Ȃ�������A���i�̏��F�E�F�̌��͂��������肷�邱�Ƃ�����܂��B ���̍X�V�ɂ��ā@�ڍׂ��������������������B �Ȃ��A���������ł́A�u�����č��E���ȓ_���v�̎x�������Ă���܂��B�ȈՓI�Ȋč��������ōs���A���邢�́A�M�Ђ��s�����ȓ_�������x������Ɩ��ł��B |
|
��Ë@�퐻���̔��ƁE�����Ɠ��̋��X�V�̃|�C���g�Ȃǂ�������Ă��܂��B |
![]()
�@
![]() ��Ë@�퐻�����o�^�̗v���@
��Ë@�퐻�����o�^�̗v���@
�u�ӔC�Z�p�ҁv�̔z�u���d�v�ȗv�f�ł��B
���@�̎���ɂ́u�\���ݔ��K���v�ւ̓K����R������܂������A���s�@�ł́A�\���ݔ���QMS�ȗ߂̍\���ݔ��֘A�K��ւ̓K�����̔��Ǝ҂�QMS�������҂ɂ��m�F����܂��B
|
�E�ӔC�Z�p�҂�z�u���� [�ӔC�Z�p���̎��i�n
|
�@
![]() �������ɂ�����QMS�ȗ߂̏��p
�������ɂ�����QMS�ȗ߂̏��p
|
QMS�ȗ���6�́i83�|84���j�ɂ��A��Ë@�퐻���Ǝ҂ɂ����Ă��K��QMS�̍\�z���K�v�ł��B �������鐻�i�ɂ��Ă��̐����Ǝ҂ōs���H���ɏƂ炵�A�K�p���邱�Ƃ��K���ł͂Ȃ�QMS�ȗ߂̗v�������ɂ��ẮA�����Ǝ҂̕i���Ǘ��ē��V�X�e���ɓK�p���Ȃ����Ƃ��ł��܂��iQMS�ȗ�85���j�B �܂��A�����̔����́A�����Ǝ҂��K��QMS���\�z���Ă��邱�Ƃ��m�F���Ȃ���Ȃ�܂���B ��Ë@��̐����ɂ����ẮA�����Ɠo�^����݂̂Ȃ炸�AQMS�̍\�z���K�v�ƂȂ�Ƃ����_�ɂ����ӂ��������B ���������ł́A�������̃��C�A�E�g�⎯�ʕ\���Ȃǂɂ��āAQMS�K���������܂��ēK�ہE���������s���Ă��܂��B |
|
��Ë@�퐻���ƎҁC��Ë@��̏��F�E�F�ɕK�{��QMS�ȗ߂ɂ��āA�ȒP�ɂ��������Ă��܂��B |
![]()
�@
![]() �V
��Ë@��̐v�J�������F�A�F�A�͏o
�V
��Ë@��̐v�J�������F�A�F�A�͏o
![]() ��Ë@�킲�Ƃɏ��F�E�F�E�͏o���K�v
��Ë@�킲�Ƃɏ��F�E�F�E�͏o���K�v
��Ë@��̔̔��̂��߂ɂ́A�Ƌ������łȂ��A��Ë@�킲�Ƃɏ��F�A�F���擾������A�͏o���s�����肵�Ȃ���Ȃ�܂���B
���F�\�����E�F�ؐ\�����E�����̔��͏��́A�v�J���֘A�̎��������f�������̂ł��B
|
�N���X�S |
���ɂ�鐻���̔����F |
|
�N���X�Q,3 |
���ɂ�鐻���̔����F |
|
�N���X�Q,3 |
������w�����������O�ҋ@�ւɂ���F���@ |
|
�N���X�P |
PMDA�ւ̐����̔��͏o |
|
�N���X�P�̂����A |
���ɂ�鐻���̔����F�@�̌�A�i���F�����E�j�͏o |
�͏o�͒�o�݂̂Ŏ葱�����������܂����A���̑��͓Ɨ��s���@�l���i��Ë@�푍���@�\(PMDA)���͔F�؋@�ւɂ��R��������܂��B
�ڍׂ́A�ʓr�����k���������B
![]()
�@
��{�v���ւ̓K��
��Ë@��́A�ł���{�I�ȗv�������ł�����{�v���i����17�N3��29�������J���ȍ�����122���j�ɓK������悤�ɐv�E�J������Ă��Ȃ���Ȃ�܂���B
���̊�{�v���ł́A�v�J����̗v�������i�d�C�I�A���w�I�A�����w�I�A�����w�I���S���Ȃǁj�ɓK�ɓK������`�ł̐v�J�����v������Ă��܂��B
��Ë@��́A��q����悤�ɁA�i�ږ��ɔF�⏳�F�A�͏o���K�v�ł����A������̈�Ë@�����{�v���ɓK�����Ă��Ȃ���Ȃ�܂���B
���ɁA�N���X�Q�ȏ�̈�Ë@��̏ꍇ�A�F�⏳�F�̐R���i�K�ŁA��{�v���ɓK�����Ă���؋������̒�o�����߂��܂��B
�܂��A�v�i�K�ŁAJIS T14971�iISO 14971�j�Ɋ�Â����X�N�}�l�W�����g�����{���Ă������Ƃ��K�v�ł��B�i���ꂪ�v�������ł̃C���v�b�g�ɂ��Ȃ�܂��j
��Ë@��̐v�J���ɓ������ẮA��{�v����JIS�K�i�Ȃǂ��悭�m�F���A���F�\���E�F�ؐ\���ŋ��߂���L�ڎ�����؋������Ȃǂ��O���ɒu���ĉ������B
![]()
�@
�w��Ǘ���Ë@��A�w�荂�x�Ǘ���Ë@��̔F��
�Ǘ���Ë@��i�N���X�Q�j�y�э��x�Ǘ���Ë@��i�N���X3�j�ɂ��ẮA�F�؊�������獐������Ă��܂��B �F�؊�ɓK��������̂��A�w��Ǘ���Ë@��A�w�荂�x�Ǘ���Ë@��Ƃ����܂��B �w��Ǘ���Ë@��E�w�荂�x�Ǘ���Ë@��̐����̔��ɂ������ẮA�����w�肷�閯�Ԃ̔F�؋@�ւɑ��āu�F�v��\�����F���擾����K�v������܂��B �F�؊�ɂ����āA��{�v���`�F�b�N���X�g�����\����Ă���A�܂��A�K�p�ƂȂ�JIS�����m������Ă��܂��̂ŁA�v�J���ɂ������Ă��Q�Ƃ��������B �F�؊�����E������̂́A�����J����b���珳�F���擾���邱�ƂɂȂ�܂��B |
�@
�A���i�̏ꍇ�̗��ӓ_
�C�O�ŗ��ʂ��Ă����Ë@���A���g�p�Ƃ���ꍇ�A���̈�Ë@�킪���{�̊�{�v����JIS���Ɋ�Â��Đv�J������Ă���킯�ł͂Ȃ����Ƃ������ł��傤�B
���̂��߁A��{�v����F�؊�ւ̓K���A�攭�i�Ƃ̔�r�����A�����E�����w�I���S���A���̑��̎��������߂Ď��{���Ȃ���Ȃ�Ȃ����Ƃ���������܂��B
�܂��A���F�i�ڂŁA�㔭��Ë@��i����^�����I�����j�A���Lj�Ë@��A�V��Ë@��ɂ����邩�̔��f�ɂ������ĕK�v�ȏ����A���{�������肵�Ă������Ƃ��K�v�ł��B
![]()
�@�@
![]() �W
�O�������Ǝғo�^�^�A����
�W
�O�������Ǝғo�^�^�A����
�����ŗ��ʂ������Ë@��̐������ŁA�v�E�傽��g���E�ŋۂ��s���O���̐������́u�O�������Ǝғo�^�v����K�v������܂��B
�܂��A������Ë@�킪�N���X�Q�ȏ�̏ꍇ�ɂ́A��Ë@��̏��F�E�F�ؐ\���̍ۂɁA���Y�O�������Ǝ҂ɑ���QMS�K�����������s���܂��̂ŁAQMS�ȗ��̊�ɂ����v���Ă���K�v������܂��B
��Ë@�퐻���̔��Ƌ����擾������́A�A�������邽�߂ɁA�V�ŏq�ׂ����F�A�F�A�͏o�̂ق��A�u�����̔��p��Ë@���A���͏��v�̒�o���K�v�ł��B
�@
![]()
�@
|
��Ë@��̐����̔��Ƃ́A��Ђ�ݗ����čs���邱�Ƃ����E�߂��܂��B �l���Ƃň�Ë@�퐻���̔��Ƌ����擾�����ꍇ�́A���i�ɂ́A�u�����v�̂ق��Ɂu�l�̐����v��\�����邱�ƂɂȂ�܂��B �l�ŋ��Ď��Ƃ�����Ă��������鎖�Ǝҗl������܂��̂ŁA�����S���ے肷����̂ł͂���܂��A �����̏ꍇ�́A �i�P�j�@��Ж��\���ɂ�����҂ւ��M���� �Ƃ������ϓ_����A��Ђ�ݗ�����邱�Ƃ������悤�ł��B �l���Ƃ̏ꍇ�A���F�͎��Ǝ�ɋA�����Ă��܂��̂ŁA���Ǝ厀�S�̏ꍇ�ɁA���ƌp���Ɏx������������ƂɂȂ�܂��B �������A�l���Ƃł���Ђł��A�����悤�ɕi���ۏE���S�Ǘ��̑̐��𐮂��Ď��Ƃ��s�����ƂɈႢ�͂���܂���B ��Аݗ��̏ڍׂ́A���������������������B
�@ |
�@
![]() 10
�����k�A���葱���̂��˗�
10
�����k�A���葱���̂��˗�
|
�@
|
|
�@�Ɋ�Â���Ë@�틖�F�\���A��Ë@�틖��Ђ̐ݗ��ȂǂɊւ��� �����k�́A���������̍s�����m�@�����@���@�������܂��B ��Ë@��Ɋւ��鎖�Ƃ̋N�Ƃ⋖�擾�����l���̕��́A�܂��́A�����k���������B �@���Ë@�틖�̊T�v�A��Ёi���Ȃ��j�ɂƂ��ĕK�v�Ȉ�Ë@��̎葱���͉����A�ȂǁA��Ђɂ��������b���������Ă��������܂��B ���������ł́A��Ë@��̐����̔��i�A���̔��j�̋��E��Ë@�퐻���Ƃ̋��\���葱���̑㗝��s��A��Ë@�퐻���̔��ƎҌ���QMS�EGVP�菇���E�L�^�l���̍쐬�A�̐��\�z�A����̉^�p�A�����č��E���ȓ_���̎x���Ȃǂ��s���Ă���܂��B ���������ł́A�����s���̎��Ǝҗl�����łȂ��A�k�C���A���k�A�֓��A�����A���A�����A�l���A��B�A���ꓙ�A�S���ŁA�V�K�Ɉ�Ë@�펖�ƂɎQ������鎖�Ǝ҂̕��̃R���T���e�B���O�������Ă��������܂����B ���u�R���T���^���g��Ёv�Ȃǂ́u�s�����m�ł͂Ȃ����v���A��Ë@��̏��F�\�����E�F�ؐ\�����A�Ƌ����\�����Ȃǂ��ƂƂ��č쐬���邱�Ƃ͖@�����ւ����Ă��܂��B���̂悤�ȃR���T���^���g��Ђ��C���^�[�l�b�g��ŎU������܂��̂ŁA�����Ӊ������B |
![]() �@���F�擾�܂ł̗���E�i�r�Q�[�V�����m�����������n
�@���F�擾�܂ł̗���E�i�r�Q�[�V�����m�����������n
![]() �@�d�b�ɂ�邲�ē��@
�@�d�b�ɂ�邲�ē��@
�\���ݕ��@�ȂǁA�������ɂ��Ă��ē��������܂��B
�ʋ�̓I�Ȏ��ē��Ɋւ���u�����k�v�́A���k�t�H�[�����A�ʒk�ɂ�邲���k�������p���������B
03-5797-5680�@�i9:30am�`�j
![]() �@�ʒk�ɂ�邲���k�@�i�\�^ZOOM�ESkype�E������
1����
10,000�~�i�Ŕ��j�j
�@�ʒk�ɂ�邲���k�@�i�\�^ZOOM�ESkype�E������
1����
10,000�~�i�Ŕ��j�j
03-5797-5680�@�i9:30am�`�j
![]() �@���[���ł̂����k�@�i�ʒk�\����ł��܂��j
�@���[���ł̂����k�@�i�ʒk�\����ł��܂��j
![]() �@�����p�́A�����Ƃ���1���Ăɂ����������ł��B
�@�����p�́A�����Ƃ���1���Ăɂ����������ł��B
![]() �@�ʋ�̓I�ȓ��e�̂�����̏ꍇ�A��̓I���ڍׂȏ���c���ł��Ȃ��l�b�g���k�̐�����A�������͈̔͂�����]�ɓY�����˂�ꍇ������܂��B
�@�ʋ�̓I�ȓ��e�̂�����̏ꍇ�A��̓I���ڍׂȏ���c���ł��Ȃ��l�b�g���k�̐�����A�������͈̔͂�����]�ɓY�����˂�ꍇ������܂��B
![]() �@�����ł̂����k�͂������������B
�@�����ł̂����k�͂������������B
![]() �@�ˋ�̃A�h���X�A�Z���A�d�b�ԍ����������̏ꍇ�A�ł��Ȃ����Ƃ�����܂��B
�@�ˋ�̃A�h���X�A�Z���A�d�b�ԍ����������̏ꍇ�A�ł��Ȃ����Ƃ�����܂��B
![]() �@�����������������l���́A���⍇���E�����k�ւ̕ԐM�A�y�сA���˗��������������ۂ̂��˗��Ɩ��̐��s�̂��߂Ɏg�p���܂��B
�@�����������������l���́A���⍇���E�����k�ւ̕ԐM�A�y�сA���˗��������������ۂ̂��˗��Ɩ��̐��s�̂��߂Ɏg�p���܂��B
�@
�@
�����k�E������t�H�[��
*�͕K�{���ڂł�
| �M���E������ |
�@ |
| *�����O | ��
��
�@ |
| *���[���A�h���X |
(�m�F�p) �@ |
| *���Z�� | ��
-
�s���{�� �s�撬���Ԓn �}���V�����E�r���� �@ |
| *���A���� | -
-
�@ |
| *�����k/ ������ |
��̓I�ɁA�����k�E��������e�����������������B ��V�z�ɂ��ẮA��̓I�Ȑ\�����e�E�R���T���e�B���O���e�Ȃǂɉ����ČʂɌ�����v���܂��B�]���܂��āA�P�ɕ�V�z�݂̂������₢���������ꍇ�A���������������˂�ꍇ���������܂��B
�@ |
| �ʒk�ɂ�邲���k ����]���� |
�E10��00���`17��30���܂ł̊Ԃł��肢���܂��B
|
�@�@ �@�@
| ��Ë@��̖\���}�j���A�� ��@�@�EQMS�����̍s�����m�ɂ���Ë@�틖�F���E���k�T�C�g
|
|||||||||||||
|
�@ |
|||||||||||||