��Ë@��̖��F�}�j���A�� �@�QMS�EGVP�����̍s�����m�ɂ���Ë@�틖�F���E���k�T�C�g

|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
�@ �@
�@
�@ |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
ver3.0�i2024�j �@
�@ �@
�ڎ� �@ 1�@��Ë@����̔��E�A�����邽�߂Ɂi�����A��Ë@��̋K���j �@ �@ �@ �@ �T�@�����́u�X�V�v�@�`���ƌp���̂��߂ɑ�Ȃ��Ɓ` �@ �U�@��Ë@��̐v�J���A���F�A�F�A�͏o �@ �@ �@ �@ ✔�@��Ë@��̐v�J���i�v���O�����j�A�������v�悵�Ă��鎖�Ǝ҂̕� ✔�@������A�����������̂���Ë@��ɊY�����邩�ǂ����s���ō����Ă��� ����ȕ��Ɍ����āA�u�ړI�������v���邽�߂̓������������܂��B �@�����}�j���A���͂������B �@
�E�l�Ⴕ���͓����̎��a���f�f�A���ÎႵ���͗\�h�Ɏg�p����邱�ƁA���� �ƒ�����̂��̂�A��t�̎w�����ʼnƒ�Ŏg�p������̂�����܂��B ��Ë@��͐l�i���ҁA��ÊW�ҁA��ʏ���ҁj�̌��N�ɉe��������̂ł��邽�߁A�u��@�@�v�i���i�A��Ë@�퓙�̕i���A�L�����y�ш��S���̊m�ۓ��Ɋւ���@���j�ɂ��A�K������Ă��܂��B �@
�@ �@ �@
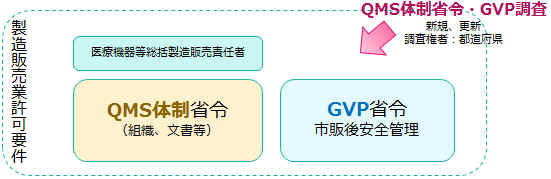
��Ë@�킪�s��ɋ��������ɂ�����A���́u�i���E�L�����E���S���v���S�ۂ���Ă��邱�Ƃ��K�{�ł��B ���̂��߂ɖ�@�@�́A���܂��܂Ȋp�x����K�������Ă��܂��B ���i�@�F���X�N�ɉ������u�N���X���ށv�Ƃ���ɉ������R�� �g�D�@�F��Ë@��̕i���E�L�����E���S�����Ǘ��ł��鎖�Ǝ҂ɑ��Ă̂݁A�����̔��������铙�A���܂��܂ȋ��E�o�^���x �Ǘ��@:��Ë@��̐����̔�����̊Ǘ���ł���QMS�ȗ߂ɂ�鐻���Ǘ��A�i���Ǘ��Ȃ� �@ ��Ë@��ɂ͑��푽�l�̂��̂�����܂��B ��ԏ����ȕ��ނ��u��ʓI�����v�Ƃ����܂��B ��ʓI���̂��Ƃɒ�`����߂��Ă��܂��B �N���X���ޕ\�͍��쌧��Web�T�C�g����i��Ë@�푍���@�\���Ŋm�F�ł��܂��B �@��
��Ë@��ɂ́A�ዾ��~�}�J�n�p�̂悤�Ȑg�߂Ȃ��̂���l�H�G�߁A�S���y�[�X���[�J�[�̂悤�Ȃ��̂܂Ŏ��ɑ��푽�l�̂��̂�����܂��B �g�p�ړI�A�g�p���@�ȂǁA���i�̎�ނɂ��A�l�̂ɑ��郊�X�N�����܂��܂ł��B ���i�̎g�p�ړI�A���ʁA�g�p���@�A�`��ȂǂɏƂ炵�A��Ë@��͈ȉ���4�́u�N���X�v�ɕ��ނ���Ă��܂�
�@ �N���X�ɉ����ċK�����قȂ�܂��B ���̎葱�����s�����Ƃ��ł���̂́u��Ë@�퐻���̔��Ƌ��Ǝ��v�ł��i�u2�v�Ō�q���܂��j�B
�@ �ŏI�I�Ɏs��ɋ��������l�����Ƃ��납��k���āA�K�v�ȗv�����߂Ă����܂��B �ǂ̂悤�Ȏg�p�ړI�A���ʂ������ǂ̂悤�ȉ��l�����i�ۑ����������j���i�𐢂̒��ɏo���Ă䂭�̂����v�悷�邱�Ƃ��̗v�ł��B �i���i�����v��Ƃ����܂��j ����ɔ����A��ʓI���́A�N���X�A���F�E�F�E�͏o�̕ʁA�ގ���Ë@��Ƃ̔�r�A���X�N�}�l�W�����g�Ȃǂ��s�����Ƃ��K�v�ƂȂ�܂��B �\���̋敪�ɉ����ď����s�ז͌ЂƂȂ�܂�����A�\���̌v���K�ɗ��Ă邱�Ƃ���Ȃ̂ł��B �����Ă��̂悤�Ȃ��Ƃ�\�������闝�R�Ƃ��Ă� �u���i���J�����܂����I ��Ë@��Ƃ��ĔF��������ł��v �Ƃ��������k�����Ȃ��Ȃ����߂ł��B ��ɕ�������A����ɑ�����t���ŔF�����悤�Ȑ��x�ł͂���܂���B ��Ë@��ɊY������ꍇ�́A��Ë@��Ƃ��Ă̋K�����܂��B �A�u�v��i�K����v��Ë@��̋K�����ӂ܂����u���i�����v�����v�J�������F�E�F�E�͏o�v�ƕ���i�߂Ȃ���Ȃ�Ȃ��̂ł��B �ꍇ�ɂ���Ă͑����̎��Ԃ��������Ƃɂ��Ȃ肩�˂܂���A�v��O�ɂ��Ђ����k����������Ǝv���܂��B �ǂ��ɑ��k����Ηǂ����s���ȏꍇ�͕��������A�����������B �����ł� �E�������I�ȂƂ��납��@�i�����̂��q�l�̑����͊F���܂��̒i�K�ł��j �E�����Ɍ��������̂�� �E������₷������� �����Ă��������Ă���܂��B �@
�@ �@ ✔�@��Ë@��̐����Ǘ��E�i���Ǘ����s�����Ђɑ��āu���v���^������ ✔�@��Ë@��̐����̂������̍H����S������ꍇ�́u��Ë@�퐻���Ɠo�^�v ✔�@QMS�ɑ������Ǘ����K�{ �@
��Ë@����s��ɏo�ׁi��s�j���邱�Ƃ��u�����̔��v�Ƃ����܂��B �����̊Ǘ��́uQMS�ȗ��AQMS�̐��ȗ߁v�uGVP�ȗ��v���ɂ��������čs���܂��B ���̊Ǘ����ł���u�̐��v�������A�ێ�����Ă����Ђɑ��Ă̂݁A�u���v���^������̂ł��B ���̂Ȃ���Ђ͈�Ë@��̐����̔����s�����Ƃ͂ł��܂���B �����̔��Ǝ҂́A�������O���ϑ����邱�Ƃ��ł��܂��B ���������̔��ӔC�ҁi��q�j�̋Ζ����鎖�Ə��i�{�Г��j������s���{���m���ɁA����\�����܂��B
�@
✔�@�g�D�A�ӔC�ƌ����̖��m�� ✔�@�Ɩ��̗���A��Ȃǂ̖��m���A������ �@ QMS��GVP�Ƃ�����ɂ̂��Ƃ��Ĉ�Ë@��̐����Ǘ��A�i���Ǘ��A���S�Ǘ����s����悤�ɁA�g�D��Ɩ��t���[�Ȃǂ𐮂��܂��B �u���ۂɂ��̑g�D�A�菇�����ɂ̂��Ƃ��Ĉ�Ë@����Ǘ�����v ���̑̐��������Ĉێ�����Ă��邱�Ƃ��A�F���܂��F���܂̂��q�l����M��������y��ɂȂ�̂ł��B �����T���v���̂悤�Ȏ菇���������Ă����悢�̂ł͂Ȃ� ✔�@��Ђ̐M�p�̑b��z�� �Ƃ�������ł�������̐����������܂��傤�B ���i�ɕs����������肻�̑��̖�肪�������Ƃ��ɉ�Ђ���邵���݂ɂ��Ȃ�܂��� ���̓y�䂪�AQMS�EGVP�ł���A�����̔��Ƌ��ł���Ƃ�����ł��傤�B �@ �@
��Ë@��̐����̔�������ӔC�҂ł��B
�i���ۏؕ���̐ӔC�҂ł��B
�i�ӂɂ͈�Ë@��A���i���͍Đ���Ó����i���̋��Ǝ҂ɂ�����3�N�̕i���Ǘ����̎����o�������߂���_�ɗ��ӂ��Ă��������B�i�����3��ɂ�����o���͌����3��̐\�����ɂ̂ݔF�߂��܂��j �@���������̔��ӔC�� ���Ƃ���Ă��܂��B ���S�m�ۋƖ��̐ӔC�҂ł��B
��1��̏ꍇ�́A���S�Ǘ����������u���A���̐ӔC�҂ł��邱�ƁA ��2��A��3��́A3�N�̌o���͋��߂��Ă��܂���B �u���̑�����ɗނ���Ɩ��v�́A�s�̌㒲���ӔC�҂̋Ɩ�����������Ƃ���Ă��܂����A�����S���ҁA��Ë@����S���҂Ƃ��Ă̋Ɩ��݂̂ł͔F�߂��Ȃ��Ƃ���Ă��܂��B �@
�@ ��������s���@���������ł́A���q�l�Ɍ��������\���ȃT�|�[�g���s���܂��B �E���q�l�ւ̃q�A�����O�@�i���O�A���l�ρA�\���w�i�A�g�D�A���̑��ʎ���j �E�ߋ�20�N�̑S���̑��l�Ȏ��Ǝҗl�̎���̒~�ρ@ �EQMS�K����������s���{���̒������ɂ��w���E�w�E��������ӂ܂������� �@ �����擾����܂ł̒i�K�́A��s�@�ɗႦ��Η����O�̒i�K�B �����ł́A�s�����m�Ƃ��āA���q�l���u�����v�i���擾�j����Ƃ��낾���łȂ� �u�����s�v���T�|�[�g���܂��B
�@
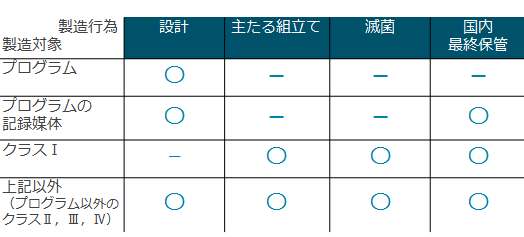
�@ �@ ✔�@��Ë@��̐����s�ׂ̂����A���̍H����S������ꍇ�́A��Ë@�퐻���Ɠo�^���� �@ �u��Ë@�퐻���Ɓv�́A��Ë@�퐻���̔��Ǝ҂���̈ϑ����Ĉ�Ë@������闧��ł��B ��Ë@��̐������̂����A���̍s�ׂ�S�����鐻�����́u��Ë@�퐻���Ɠo�^�v���܂��i�o�^�������������u�o�^�������v�Ƃ����܂��j �o�^���邽�߂ɂ́A�v�������ӔC�ҁu�ӔC�Z�p���v�����n�Ǘ��ł���悤�z�u���邱�Ƃ��K�v�ł��B ���������O���ɂ���ꍇ�́u�O�������Ǝғo�^�v��\�����܂��B�ӔC�Z�p�҂͋��߂��܂���B �@ �Z�����Ă���H����S�����鐻�����́A��Ë@�퐻���Ɠo�^���K�v�ł�
�@ Q�@�\���ݔ��͂ǂ̂悤�ɐR������܂����@�L���v���͂���܂��� ���̂悤�Ȃ�����������������Ƃ�����܂��B ���͈�Ë@�퐻���Ɠo�^�̐R���ł́A�\���ݔ����Ó��Ȃ��̂ł��邩�ǂ����A�L���͏\�����Ƃ��������Ƃ͗v���ɂȂ��Ă��܂���B �\���ݔ��̑Ó����ɂ��ẮA���������Ë@��ɉ����Đ����Ǝҁi����̔��Ǝҁj�����f���܂��B �i�ꕔ�A�K�肪���O����鐻�i������܂��j �@ �����Ǝ҂́AQMS�ȗ߂Ɋ�Â��������̕i���}�l�W�����g�V�X�e���iQMS�j���\�z����K�v������AQMS�̑̐��\�z�͐����̔��Ǝ҂ɂ����Ҋč���R���@�ւɂ��R���̑ΏۂƂȂ�܂��B �\���ݔ��͓s���{���̐R�����鋖�v���ł͂Ȃ����̂́AQMS�ȗ߂̊֘A����K��Ɋ�Â��K�ɉ^�p�Ǘ�����Ȃ���Ȃ�܂���B �@
�u�ӔC�Z�p�ҁv�͈�Ë@��̐����������n�ɊǗ�����ӔC�҂ł��B
�@ �i�ڍׂ�4����������� >>�j �@
�@ �@ �@
�@
��s�@�ɂ��Ƃ��ĕ\�����Ă݂܂����B ���ꂩ�狖����낤�Ƃ����i�K�́A��s�@�̐��������Ă���i�K�B ���ꂪ �E�g�D�@�i����A�ӔC�ҁj �E�Ɩ��t���[�i�v���Z�X�j�̖��m���@����d���̊�Ǝ菇 �E�^�p���邽�߂̒m���̃C���v�b�g �Ȃ̂ł��B 20�N�ȏ�A�͂��߂Ĉ�Ë@�����舵�����q�l�̎x�������Ă��āA �厖�Ȃ̂́u�̐��v���������萮���邱�Ƃ��Ǝ������Ă��܂��B �T���v���̂悤�Ȏ菇�����ۂ�Ƃ����Ă��������ł͈Ӗ����Ȃ��A ������K�v�ȏꍇ�͐v���I�m�Ɏ��{���邱�ƂŁA�ނ���Ǘ��̐��������Ă��邱�Ƃ������邱�Ƃ�����ł��傤�B �@ ��������s���@���������ł́A�u������݂����āv �P�Ɂu�\����s�v����̂ł͂Ȃ� �u�����`�����`�����s�܂ł��T�|�[�g�v���Ă��܂��B �@
�s���{���ł́A�R�����ɗp�ӂ���Ă���菇�����K�ł��邩�ǂ����A���̓��e�����Ǝ҂��������āA�K�ɉ^�p�ł�����ǂ����Ȃǂ��A���n�����Ŕ��f���܂��B �\���i�K�ł͂������܂����Ƃ̓X�^�[�g���Ă��܂��A��舵����Ë@��A��Ђ̑g�D�A�̘H���ɉ����A���擾��K�ɉ^�p�ł���悤�Ȏ菇���E�g�D�ɂȂ��Ă��邱�Ƃ��A�V�K�\�����̌��ł��B ���������̂����k�E���˗��Č��̒��ɂ́A����\�������ꂽ���Ǝ҂̂����ŁAQMS�EGVP�菇�����s�K���Ȃ��߂ɂȂ��Ȃ�������OK�����炦���A���擾���Ȃ��Ȃ��ł��Ȃ������������܂��B �Ȃ��Ȃ�OK���ł܂���ƁA�����\�肵�������Ɏ��Ƃ��X�^�[�g�ł��Ȃ��悤�Ȃ��Ƃɂ��Ȃ肩�˂܂���B �ł�����A�����i�K�ŁA����P����ʂ��āA��@�@���F��m��AQMS�EGVP�̗v���������悭�������āA �T�v���C���[�E�������Ƃ̊W�A������Ë@��̓����A�Г��g�D���܂��A���̂���菇�������邱�Ƃ�������K�{���Ƃ�����ł��傤�B ���������ł́A�V�K�\���⋖�X�V���ɍۂ��A����P���̈�Ƃ��āA����/���S���җl�ɖ@�EQMS�EGVP�������������������ƂɎ���u��������P���i�Г�����j�̋@���݂��Ă���܂��B �@
�����Зl�i�w�Ёj�̎���ł��i���`���ɒ�G���Ȃ��悤�ɃA�����W���Ă��܂��j �Г���QMS�̕������쐬���A�����s�ɋ��\�������Ƃ���A�ЂƂ܂��u�K���v�Ɣ��f���ꋖ�邱�Ƃ��ł��܂����B�����̓l�b�g��ɂ���T���v�������p���܂����B ����S���Ǝv������A�����ł͂���܂���ł����B �����̔��Ƌ����擾������ɁA��Ë@��̔F�ؐ\�����s���AQMS�ȗ߂ɓK�����Ă��邩�ǂ����̒������邱�ƂɂȂ�܂����B �����ŁA�Ȃ��40�����́u�s�K���v���w�E���ꂽ�̂ł��B �Ȃ������������Ƃ��N�����̂ł��傤���B �����s����������͈͂͌���I�ł���A�܂��A�ڍׂł͂���܂���B ���R�́A�������쐬����ۂɃT���v���𗘗p�������Ƃɂ���܂����B �@
���̂悤�Ȃ��Ƃ�h�����߂ɂ� ✔�@���̎��_ ✔�@���\�����s�ł���@�I�Ȍ��� ✔�@QMS�̗��� ✔�@�����T�|�[�g����̐� ���K�v�ł��傤�B ������������āA �EQMS�̐��\�z �E���\�� �EQMS�̉^�p�@�i�����x���j �E�F�ؐ\�� �EQMS�K���������̎�R ���g�[�^���Ŏx���ł���s�����m�Ɉ˗����Ă����������Ƃ������߂��܂��B �@
�@ �@ ✔�@�pMS�����ɑ������^�p���s�� ✔�@�^�p�̑��k����m�ۂ��� �@ ��Ë@��̎��Ƃ��p������ɂ� �E��Ë@�퐻���̔��Ƌ��̍X�V �E��Ë@�퐻���Ɠo�^�̍X�V �E��Ë@��̏��F�A�F�̈ێ��@�iQMS�K���w��������K�����X�V�j �@ ��������Ɉӎ����Ă������Ƃ��̗v�ł��B �Ȃ��Ȃ�� �E�ߋ�5�N�Ԃ̉^�p���������蒲������� ����Ȃ̂ł��B �@ �V�K�ɋ��\�������Ƃ��́A�����������璲���S���҂��D�������ŁA�������苖�ɂȂ�����������܂���B���������ꂪ���X�ɂ��ė��Ƃ����ɂȂ�܂��B 5�N��̐����̔��Ƌ��X�V�̍ۂɂ́A�uQMS�̐����ێ�����Ă��邱�Ɓv���������蒲������܂��BQMS�̐��̈ێ������v��������ł��B �EQMS������GVP�菇�����K���́A�L���ȏȗ߂��K��������̂ł��邩 �EQMS�����ɂ̂��Ƃ��āA�i�����j����Ǘ��ēҏƍ��܂��K�Ɏ��{����Ă��邩 ���̂悤�ɁA�`���ʂƉ^�p�ʂ��A�����ƋL�^�A�����ł̃q�A�����O��ʂ��Ċm�F����܂��B 5�N�ԓK�ɉ^�p����Ă��Ȃ��ƁA�����ōX�V���ł��Ȃ��P�[�X���o�Ă���̂ł��B �@
��Ë@��̏��F�E�F���ێ����A�܂���Ë@�퐻���̔��Ƌ�����Ɠo�^���X�V���邽�߂ɂ́AQMS�AGVP�̐��̐�����QMS�ȗ߂ւ̓K���A�i�ڂ��Ƃ̋L�ڐ����Ȃǂ̏��葱�����K�v�ł��B ���߂Ă̋��擾���ƈقȂ�A�O��̍X�V�ȍ~��QMS�EGVP���K�ɉ^�p����Ă������ǂ���������܂��B �R���i�K��QMS�EGVP�̉^�p���\���łȂ��A�Ƃ����ɂȂ�܂��ƁA�ꍇ�ɂ���Ă͋��������߂��Ă��X�V�ł��Ȃ�������A���i�̏��F�E�F�̌��͂��������肷�邱�Ƃ�����܂��B ���̍X�V�ɂ��ā@�ڍׂ��������������������B �Ȃ��A���������ł́A�u�����č��E���ȓ_���v�̎x�������Ă���܂��B�ȈՓI�Ȋč��������ōs���A���邢�́A�M�Ђ��s�����ȓ_�������x������Ɩ��ł��B
�@ �@
��Ë@��̔̔��̂��߂ɂ́A�Ƌ������łȂ��A��Ë@�킲�Ƃɏ��F�A�F���擾������A�͏o���s�����肵�Ȃ���Ȃ�܂���B ���F�\�����E�F�ؐ\�����E�����̔��͏��́A�v�J���֘A�̎��������f�������̂ł��B
�͏o�͒�o�݂̂Ŏ葱�����������܂����A���̑��͓Ɨ��s���@�l���i��Ë@�푍���@�\(PMDA)���͔F�؋@�ւɂ��R��������܂��B �ڍׂ́A�ʓr�����k���������B
�@
|

 �����X�A���ˊ�A��×p�K�[�[�Ȃǂ���A��pX���f�f���u�Ȃǂ̈�p�d�C�@��A���͋@��܂ŁA��Ë@��ɂ́A���܂��܂Ȍ`��A�p�r�̂��̂�����܂��B
�����X�A���ˊ�A��×p�K�[�[�Ȃǂ���A��pX���f�f���u�Ȃǂ̈�p�d�C�@��A���͋@��܂ŁA��Ë@��ɂ́A���܂��܂Ȍ`��A�p�r�̂��̂�����܂��B


�Ǘ���Ë@��i�N���X�Q�j�y�э��x�Ǘ���Ë@��i�N���X3�j�ɂ��ẮA�F�؊�������獐������Ă��܂��B �F�؊�ɓK��������̂��A�w��Ǘ���Ë@��A�w�荂�x�Ǘ���Ë@��Ƃ����܂��B �w��Ǘ���Ë@��E�w�荂�x�Ǘ���Ë@��̐����̔��ɂ������ẮA�����w�肷�閯�Ԃ̔F�؋@�ւɑ��āu�F�v��\�����F���擾����K�v������܂��B �F�؊�ɂ����āA��{�v���`�F�b�N���X�g�����\����Ă���A�܂��A�K�p�ƂȂ�JIS�����m������Ă��܂��̂ŁA�v�J���ɂ������Ă��Q�Ƃ��������B �F�؊�����E������̂́A�����J����b���珳�F���擾���邱�ƂɂȂ�܂��B |
�@
 �A���i�̏ꍇ�̗��ӓ_
�A���i�̏ꍇ�̗��ӓ_
�C�O�ŗ��ʂ��Ă����Ë@���A���g�p�Ƃ���ꍇ�A���̈�Ë@�킪���{�̊�{�v����JIS���Ɋ�Â��Đv�J������Ă���킯�ł͂Ȃ����Ƃ������ł��傤�B
���̂��߁A��{�v����F�؊�ւ̓K���A�攭�i�Ƃ̔�r�����A�����E�����w�I���S���A���̑��̎��������߂Ď��{���Ȃ���Ȃ�Ȃ����Ƃ���������܂��B
�܂��A���F�i�ڂŁA�㔭��Ë@��i����^�����I�����j�A���Lj�Ë@��A�V��Ë@��ɂ����邩�̔��f�ɂ������ĕK�v�ȏ����A���{�������肵�Ă������Ƃ��K�v�ł��B
![]()
�@�@
![]()
�@
![]() �V
��Ђ̐ݗ�
�V
��Ђ̐ݗ�
|
��Ë@��̐����̔��Ƃ́A��Ђ�ݗ����čs���邱�Ƃ����E�߂��܂��B �l���Ƃň�Ë@�퐻���̔��Ƌ����擾�����ꍇ�́A���i�ɂ́A�u�����v�̂ق��Ɂu�l�̐����v��\�����邱�ƂɂȂ�܂��B �l�ŋ��Ď��Ƃ�����Ă��������鎖�Ǝҗl������܂��̂ŁA�����S���ے肷����̂ł͂���܂��A �����̏ꍇ�́A �i�P�j�@��Ж��\���ɂ�����҂ւ��M���� �Ƃ������ϓ_����A��Ђ�ݗ�����邱�Ƃ������悤�ł��B �l���Ƃ̏ꍇ�A���F�͎��Ǝ�ɋA�����Ă��܂��̂ŁA���Ǝ厀�S�̏ꍇ�ɁA���ƌp���Ɏx������������ƂɂȂ�܂��B �������A�l���Ƃł���Ђł��A�����悤�ɕi���ۏE���S�Ǘ��̑̐��𐮂��Ď��Ƃ��s�����ƂɈႢ�͂���܂���B ��Аݗ��̏ڍׂ́A���������������������B
�@ |
�@
|
�@
|
|
�@�Ɋ�Â���Ë@�틖�F�\���A��Ë@�틖��Ђ̐ݗ��ȂǂɊւ��� �����k�́A���������̍s�����m�@�����@���@�������܂��B ��Ë@��Ɋւ��鎖�Ƃ̋N�Ƃ⋖�擾�����l���̕��́A�܂��́A�����k���������B �@���Ë@�틖�̊T�v�A��Ёi���Ȃ��j�ɂƂ��ĕK�v�Ȉ�Ë@��̎葱���͉����A�ȂǁA��Ђɂ��������b���������Ă��������܂��B ���������ł́A��Ë@��̐����̔��i�A���̔��j�̋��E��Ë@�퐻���Ƃ̋��\���葱���̑㗝��s��A��Ë@�퐻���̔��ƎҌ���QMS�EGVP�菇���E�L�^�l���̍쐬�A�̐��\�z�A����̉^�p�A�����č��E���ȓ_���̎x���Ȃǂ��s���Ă���܂��B ���������ł́A�����s���̎��Ǝҗl�����łȂ��A�k�C���A���k�A�֓��A�����A���A�����A�l���A��B�A���ꓙ�A�S���ŁA�V�K�Ɉ�Ë@�펖�ƂɎQ������鎖�Ǝ҂̕��̃R���T���e�B���O�������Ă��������܂����B ���u�R���T���^���g��Ёv�Ȃǂ́u�s�����m�ł͂Ȃ����v���A��Ë@��̏��F�\�����E�F�ؐ\�����A�Ƌ����\�����Ȃǂ��ƂƂ��č쐬���邱�Ƃ͖@�����ւ����Ă��܂��B���̂悤�ȃR���T���^���g��Ђ��C���^�[�l�b�g��ŎU������܂��̂ŁA�����Ӊ������B |

![]() �@���F�擾�܂ł̗���E�i�r�Q�[�V�����m�����������n
�@���F�擾�܂ł̗���E�i�r�Q�[�V�����m�����������n
![]() �@�d�b�ɂ�邲�ē��@
�@�d�b�ɂ�邲�ē��@
�\���ݕ��@�ȂǁA�������ɂ��Ă��ē��������܂��B
�ʋ�̓I�Ȏ��ē��Ɋւ���u�����k�v�́A���k�t�H�[�����A�ʒk�ɂ�邲���k�������p���������B
03-5797-5680�@�i9:30am�`�j
![]() �@�ʒk�ɂ�邲���k�@�i�\�^ZOOM�ESkype�E������
1����
10,000�~�i�Ŕ��j�j
�@�ʒk�ɂ�邲���k�@�i�\�^ZOOM�ESkype�E������
1����
10,000�~�i�Ŕ��j�j
03-5797-5680�@�i9:30am�`�j
![]() �@���[���ł̂����k�@�i�ʒk�\����ł��܂��j
�@���[���ł̂����k�@�i�ʒk�\����ł��܂��j
![]() �@�����p�́A�����Ƃ���1���Ăɂ����������ł��B
�@�����p�́A�����Ƃ���1���Ăɂ����������ł��B
![]() �@�ʋ�̓I�ȓ��e�̂�����̏ꍇ�A��̓I���ڍׂȏ���c���ł��Ȃ��l�b�g���k�̐�����A�������͈̔͂�����]�ɓY�����˂�ꍇ������܂��B
�@�ʋ�̓I�ȓ��e�̂�����̏ꍇ�A��̓I���ڍׂȏ���c���ł��Ȃ��l�b�g���k�̐�����A�������͈̔͂�����]�ɓY�����˂�ꍇ������܂��B
![]() �@�����ł̂����k�͂������������B
�@�����ł̂����k�͂������������B
![]() �@�ˋ�̃A�h���X�A�Z���A�d�b�ԍ����������̏ꍇ�A�ł��Ȃ����Ƃ�����܂��B
�@�ˋ�̃A�h���X�A�Z���A�d�b�ԍ����������̏ꍇ�A�ł��Ȃ����Ƃ�����܂��B
![]() �@�����������������l���́A���⍇���E�����k�ւ̕ԐM�A�y�сA���˗��������������ۂ̂��˗��Ɩ��̐��s�̂��߂Ɏg�p���܂��B
�@�����������������l���́A���⍇���E�����k�ւ̕ԐM�A�y�сA���˗��������������ۂ̂��˗��Ɩ��̐��s�̂��߂Ɏg�p���܂��B
�@
�@
�����k�E������t�H�[��
*�͕K�{���ڂł�
| �M���E������ |
�@ |
| *�����O | ��
��
�@ |
| *���[���A�h���X |
(�m�F�p) �@ |
| *���Z�� | ��
-
�s���{�� �s�撬���Ԓn �}���V�����E�r���� �@ |
| *���A���� | -
-
�@ |
| *�����k/ ������ |
��̓I�ɁA�����k�E��������e�����������������B ��V�z�ɂ��ẮA��̓I�Ȑ\�����e�E�R���T���e�B���O���e�Ȃǂɉ����ČʂɌ�����v���܂��B�]���܂��āA�P�ɕ�V�z�݂̂������₢���������ꍇ�A���������������˂�ꍇ���������܂��B
�@ |
| �ʒk�ɂ�邲���k ����]���� |
�E10��00���`17��30���܂ł̊Ԃł��肢���܂��B
|
�@�@ �@�@
| ��Ë@��̖\���}�j���A�� ��@�@�EQMS�����̍s�����m�ɂ���Ë@�틖�F���E���k�T�C�g
|
|||||||||||||
|
�@ |
|||||||||||||