QMS�ȗߓK�p�̈�Ë@�� �b ��Ë@��̖@���F�¥GQP�����̍s�����m ��������s���@��������

|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
�@ �@
�@
�@ �@ |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
�@
�@��Ë@��̐����̔��ɗv�������QMS�EGVP�̐��Ɖ^�p
�����̔��Ǝ҂��琻���ϑ����Ă����Ë@�퐻���Ǝ҂ɂ����Ă��A�K�Ȑ����Ǘ��E�i���Ǘ��̐�������K�v������܂��B�@
��Ë@��̏��F�E�F�̈ێ��̗v���ɂ��Ȃ��Ă��܂��B �܂��A��Ë@��̐����̎�������鐻���Ǝ҂��AQMS�ȗ߂��K�p�ɂȂ�܂��B��Ë@�퐻���Ɠo�^����݂̂ł͂Ȃ��AQMS�ȗ߂ւ̑Ή����܂߂āA����̌v��Ə�����i�߂�K�v������܂�
�@ QMS��
Quality Management System
�i���Ǘ��ēV�X�e���̗��ł��B ��Ë@��̐����̔��Ǝ҂��琻���ϑ������Ë@�퐻���Ǝ҂��A�K�ȕi���Ǘ��ēV�X�e�����\�z������ɏ]���ċƖ����s��Ȃ���Ȃ�܂���B
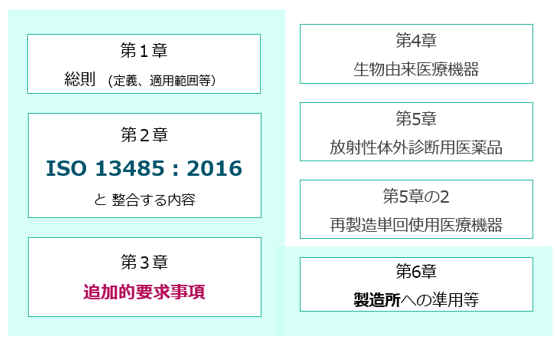
�@ �@ QMS�ȗ߂�QMS�i�i���Ǘ��ēV�X�e���j�̑g�D�╶���A�^�p�ɂ��Ē�߂���ł��B ��2021�N�{�s��QMS�ȗ߂�ISO13485:2016�Ɛ���������e�ƂȂ��Ă��܂��B
�@ 2021�N3��26���ɉ���QMS�ȗ߂��{�s����Ă��܂��B �]�O�̏ȗ߂��o�ߑ[�u�Ƃ���2024�N3��25���܂ł͗L���ł��B �܂��A14971�̉����iJIS T14971:2020�j�ւ̑Ή��i2023�N9��30���܂Łj�A���[�U�r���e�B�K�iJIS T62366-1:2019�ւ̑Ή������K�v�ƂȂ�܂��̂ŁA�����ӂ��������B ��QMS�ȗ߂̉����ւ̑Ή��̏����E�x�����ɂ��ẮA�����炩�����₢���킹���������B
�@ GVP��
Good Vigilance Practice
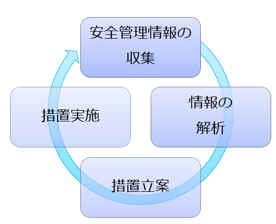
�̗��ŁA�����̔���̈��S�Ǘ��̊�ł��B ���̖̂��R�h�~�ƁA��蔭�����̐v���ȑΉ��̂��߂̎d�g�݂Ƃ�����ł��傤�B GVP�ȗ߂ւ̓K���́A�����̔��Ƌ��̗v���ł���A�s���{���������̔��Ƌ��̐V�K�̐\�������Ƃ���X�V�O�ɁA���Ǝ҂ɑ��Ē��������{���܂��B �܂��AQMS�ȗ߂̒��ł��A���̎�t�`�Ή����AGVP�Ɋւ��K�肪���邽�߁AQMS�K���������̒��ł�GVP�̎菇����^�p�ɂ��Ē�������邱�Ƃ�����܂��B
�@ �@ �@ �@
�N���X�Q~�S�̈�Ë@��́A���F�E�F�ؗv���Ƃ���QMS�ȗ߂ɓK�����邱�Ƃ��K�v�ł��B �i���\�Łu�K�{�v�Ƃ���̂́A�V�K���F�E�F�؎���A����������ɂ�鏳�F�E�F�؈ێ��̂��߂ɁA�K�������߂��邱�Ƃ��Ӗ����Ă��܂��j �@
�@ ���@�N���X�P�̈�Ë@��ɂ��ẮAQMS�K���������͂���܂���B �@ �@ �N���X�P�̈�Ë@��̂����A�u�����ʈ�Ë@���v�ƍ����w�肵����Ë@��i��ʓI���̂��ƂɎw��j�́AQMS�ȗ߂̂����A�ꕔ�̗v�����K�p�ɂȂ�܂���B �܂��A�����ʈ�Ë@��݂̂��̔����鐻���̔��Ǝ҂̂��Ƃ��A�����O����Ë@�퐻���̔��Ǝ҂Ƃ����A�K�p�ɂȂ�Ȃ���������������A�ȗ������ꂽ�Ǘ��̐����F�߂��Ă��܂��B QMS�ȗ߂Ɋ�Â��ĕi���}�j���A����֘A�菇�����쐬���邤���ŁA�����ʈ�Ë@��������ꍇ��A�����O��ɊY������ꍇ�ɂ́A����ɉ����������쐬���K�v�ɂȂ�܂��B �@ ���@�����ʈ�Ë@��ł͂Ȃ��N���X�P�̈�Ë@��́A�������������������>> ���@�N���X�P�̈�Ë@��́A30-36����2�i�v�J���j�͓K�p�ɂȂ�܂���B �@ �@
�@�@
���ꂩ���Ë@��̋Ƌ����擾���A���i�̓͏o��������F�E���F���擾�����肵�悤�Ƃ������Ǝ҂̕���A �@
�܂��́AQMS�̐��ȗ߁EQMS�ȗ߂�GVP�ȗ߂ŁA�ǂ̂悤�Ȏd�������߂��Ă���̂���m��܂��傤�B �@ �����āA
�@�������������m�ɂ��܂��B �@�i�����ł��A�T�|�[�g�̂��˗��������������ꍇ�A�����̃X�e�b�v�����s���܂��j
QMS�ȗ߂�GVP�ȗ߂ł́A�Ǘ��ēҁA�iQMS�́j�Ǘ��ӔC�ҁA���������̔��ӔC�ҁA�����i���Ɩ��^�c�ӔC�ҁA���S�Ǘ��ӔC���iGVP�j�Ȃǂ��o�ꂵ�܂��B ���ꂼ��v��������A�ȗ߂Ɋ�Â�������S���Ă��܂��B �����擾���邽�߂ɂ����v�������l�𑵂���悢�̂ł͂Ȃ��A���ۂ̉�Ђ̎w�����ߌn���A����Ԃ̘A�g�������Ă��āA�ǂ̕�����i������E���S����ɂ��邩�Ƃ��������Ƃ��������Ă��������B �d�������邤���őg�D�ƂƂ��ɏd�v�ȂȂ̂��u�����v�ł��B �@ �@
��Ë@��̐����Ǘ��E�i���Ǘ��A�����̔�����S�Ǘ����s�����߂ɂ́A���̎菇�┻�f��m�������e��̕������߁A����Ɋ�Â��ċƖ����s�����Ƃ��K�v�ł��B QMS�̋Ɩ��̊�{�������i���Ǘ��ēV�X�e������i�i���}�j���A���j�Ƃ����A���̉��ɁA�ʂ̋Ɩ��̎菇�Ȃǂ��߂����ʕ������߂܂��B ��3���Ë@�퐻���̔��Ǝ҂̏ꍇ�AGVP�̎菇���i�����̔�����S�Ǘ��Ɩ��菇���j�̍쐬�͋`���t�����Ă��܂��A�Ɩ���K�ɍs����̐��𐮂��邤���ŁA�菇���܂�H������ɗނ��镶���𐮂��邱�Ƃ͎�����K�v�ƍl�����܂��B �@
�@ �@
�������������ނ́A�ꕔ�͂ЂȌ`�̂悤�Ȃ��̂��C���^�[�l�b�g��ɃA�b�v���[�h����Ă��܂��B �ł́A��Ë@��̐����̔��Ƌ��擾�̏����i�K�ŁA�������_�E�����[�h���Ă����Ă����悢�̂ł��傤���B
�@ �i�����ł͂��̂悤�Ȏ���𐔑������Ă��܂����B �@ �@
�ł�����A�����ЂȌ`���_�E�����[�h���Ă����Ă������Ƃ��ړI�ł͂Ȃ����ƁA��ЂɌ��������d���̎d�����`��邱�Ƃ��d�v���Ƃ����_�ɗ��ӂ��Ă������������Ǝv���܂��B �i�����ŃT�|�[�g�����Ă��������ۂɂ́A�ō����A�q�A�����O���o�āA�����̃I���W�i���̕����Ƃ��č쐬�܂��͋M�Ђł̍쐬���T�|�[�g�����Ă��������Ă���܂��B�ЂȌ`���̂܂܂Ƃ������Ƃ͂���܂���j �@ �����擾���邱�Ƃ́A�ړI�i�S�[���j�ł͂Ȃ��A����͎��Ǝ҂Ƃ��ẴX�^�[�g�Ȃ̂ł��B ���Ђ̑g�D�A�ϑ���̗L���A�戵���i�̐v�J������o�ׂ܂ł̍H���A���i�̐����Ȃǂ����Ă��č쐬���܂��傤�B �@ �����āA�菇���́A��x�������I���łȂ��A�����č��E���ȓ_���Ȃǂ̋@������p���ĉ��P���Ă䂫�܂��傤�B �@ �F�ؕi�ڂł���A�F�؎擾�ȍ~���N�u�T�[�x�C�����X�v�Ƃ����R������5�N�ڂ̒���������s���܂��B �@
�@
�悭���鑨�����́u���F�\���́A���ނ𐮂��Ė����ɏo���悢�v�Ƃ����u���ނ��肫�v�u�i��ߐ��́j�葱���v�Ƃ����l�����ł��B �����A�����ɁAQMS��GVP�̑Ή��A�����̔��Ƌ�����Ɠo�^�A���i�̎葱���Ȃǂ����܂��܂ȗv�f���W���܂��̂ŁA���̂悤�ȑ������͐������Ƃ͌����Ȃ��ł��傤�B ��Ë@����̔�������A���F��F���ێ������肷�邽�߂ɂ́A�葱�����ܘ_��ł����i�����Ă��̂��߂ɗl�X�ȏ������������܂����j�A �����d�v�Ȃ̂́A���������葱���i�@�K���j���܂߂đΉ��ł���A��@�@�Ή��͂���Ђ������A�@�ᔽ��ԂȂǂɂȂ�Ȃ��悤�ɊǗ����Ă䂭���Ƃł͂Ȃ��ł��傤���B �܂��͌����c�����A�v�Ή������m�����A����ɉ������v��𗧂Ă邱�Ƃł��B �@
�@ �����E�I�m�ȑΉ��̊̂́A�ŏ��Ɍ���c���E�v�Ή��������m���E���{�v��̗��Ăɂ��邩������܂���B �������̃T�|�[�g���e�́A���q�l�ɂ��A�܂����i�̏ɂ���Ă��A�قȂ�܂��B �@
�@
QMS�ȗߑ�2�͂�ISO13485:2003�Ɠ��l�̓��e�ł��B
�@
�C���^�[�l�b�g��ɍڂ��Ă���s���{�����̐��`�̂܂ܐ����̔��Ƌ��\��������ƁA�s���{���ł͖��Ȃ��Ƃ��ĔF�߂邱�Ƃ�����܂��B �������A���ۂɂ�����^�p����̂͊F�l�̉�Ђł� ���擾��AQMS�̒�������ہi�F�؋@�֓������{�j�A��Ђ̎���Ƃ̘������傫���A�^�p������Ă��Ȃ��AQMS�ȗ߂̗��������Ă��Ȃ��Ȃǂ̗��R�ɂ��A�������̎w�E����P�[�X���U������܂��B ����ɂ���Ë@��̔F����܂ł̃X�P�W���[����傫���������Ȃ���Ȃ�Ȃ��Ȃ邱�Ƃ�����܂��B �����ɂ��A���̂悤�Ȏ��_�ŏ��߂Ă����k�����������Ⴊ����܂��B �܂��A���Ђ����Ŏ��g�ނ��Ǝ��̂͌o���̒~�ςɂȂ�ʂ͂���̂ł����A��O�҂̖ڂ�����Ȃ����߂ɋ��F�擾��̃��X�N�ɂ��Č����Ƃ��Ă���P�[�X������܂��B ����F�̎擾�͖ړI�ł͂Ȃ��A�X�^�[�g�n�_�ɗ��ɂ����܂��� ���Ǝ҂Ƃ��Ĉ�Ë@��̔F�ؓ����ێ����ēK�ɖ@�߂����炵�Đ����̔������邽�߂ɂ́A�v��i�K����O���̖ڂ���ꏕ���E�x�����邱�Ƃ������߂��܂��B �悢�s�����m��x���҂ł���A���̎x���̌��ʂ���Ђ̖��`�̍��Y�ƂȂ�ł��傤�B �@
�@ �@
��Ë@��́A���p�ҁA���ҁA��ÊW�ғ��̌��N��Q�ɒ�������\����������̂ł��B �ł�����A��Ë@��̐����̔��ɂ������ẮA���̎��Ǝ҂ɐ����Ǘ��E�i���Ǘ��A���S�Ǘ����ł���̐������ƁA��ɂ����������^�p�Ǘ������߂��Ă��܂��B ���Ǝ҂́A�e��菇�������쐬������A�Г�����������肵�āA��背�x���ȏ�̋Ɩ����ł���悤�}��̂ł��B
�@�߂ɏ]�����K�Ȏ菇���Ɋ�Â��^�p�́A�Ɩ��̎������コ����ƂƂ��ɁA��Ƃ̖@�I���X�N�����炩���ߒጸ���Ă����ł��傤�B ������A��Ë@��ɋN������Ǝv����g���u���i�@�I�Ȗ��j���������ꍇ�ɂ́A��Ђ��@�K�������炵�Ή����Ă������Ƃ��������ׂɂȂ邩������܂���B �����������ϓ_������A���f���菇�����̂܂܂ł��邱�Ƃ͂������ă��X�L�[���Ƃ�����ł��傤�B �@
�@ ��Ë@��̐����̔��Ǝ҂���Ǝ҂́A�����̖@�I���X�N��ጸ�����������{���邱�Ƃ��K�v�ł��B ��
�s�����m�́A���������@�I���X�N�����Ă��Ȃ���A�e��菇�����쐬���܂��B �@
�@ �@
�@
�\���ݕ��@�ȂǁA�������ɂ��Ă��ē��������܂��B 03-5797-5680�@�i9:30am�`�j
03-5797-5680�@�i9:30am�`�j
�����k�E������t�H�[��*�͕K�{���ڂł�
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ | �@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||


| ��Ë@��A��×p��̋��F�\���AGQP�EGVP�����̍s�����m
|
|||||||||||||
|
�@ |
|||||||||||||
�@